Explorar las categorías
Explora
Fiverr Pro
Español
$
USD
Experto en bioinformática profesional en diseño de fármacos y análisis de datos
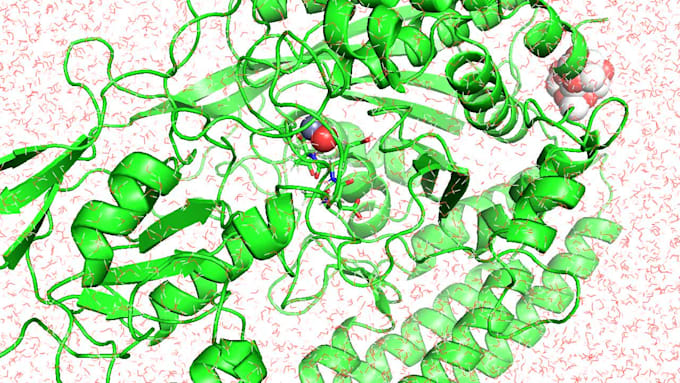
Ofrezco servicios profesionales de simulaciones de dinámica molecular (MD) basadas en GROMACS para sistemas de proteína-ligando, proteína-ligando-ion y proteína-ligando-nanopartícula o cúmulo. Puedo ayudar con la configuración del sistema, ejecución de la simulación, análisis de trayectorias e interpretación científica.
Los servicios incluyen:
La duración de la simulación y los precios se personalizan según la complejidad del sistema y los requisitos computacionales.
Por favor, contáctame antes de hacer un pedido para discutir los requisitos de tu proyecto.



Dominio:
Otros
Experiencia:
Análisis predictivo
•
Otros
Lenguaje de programación:
Python
•
R
•
Otros
Herramientas:
Jupyter Notebook
•
Otros
Tecnología:
Python
•
R
•
Excel
•
Otros
Modelos y métodos:
Aprendizaje automático
•
Deep learning
•
Otros