Explorar las categorías
Explora
Fiverr Pro
Español
$
USD
Experto en descubrimiento de fármacos computacional, bioinformática y entrada de datos
¡Bienvenido a mi servicio profesional de Bioinformática y Biología Computacional!
Si buscas investigación in silico de alta calidad, docking molecular y simulaciones avanzadas de dinámica molecular (MD), estás en el lugar correcto. Ofrezco datos computacionales precisos y listos para publicación para tu investigación.
Lo que ofrezco:
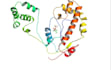
Docking molecular: receptor-ligando, proteína-proteína y cribado virtual.
Simulaciones MD: simulaciones de larga duración (GROMACS/LAMMPS) para evaluar la estabilidad estructural.
Análisis post-simulación: RMSD, RMSF, radio de gyración (Rg) y análisis de enlaces de hidrógeno.
Visualización de datos: gráficos de interacción de alta resolución, estructuras 3D y gráficos de calidad para publicación.
Herramientas que utilizo:
GROMACS, LAMMPS, AutoDock Vina, PyMOL, VMD y Python (para análisis de datos personalizados).
¿Por qué elegirme?
Investigador computacional dedicado.
Datos científicos de alta calidad y reproducibles.
Entrega puntual y informes técnicos estructurados.
Por favor, contáctame antes de hacer un pedido para discutir el flujo de trabajo de tu proyecto.



Traducción automática
¿Qué software/herramientas utilizas para simulaciones de MD y acoplamiento?
Principalmente uso GROMACS y LAMMPS para simulaciones de dinámica molecular. Para el acoplamiento molecular, utilizo AutoDock Vina, y para visualización, empleo PyMOL y VMD.
¿Necesito proporcionar las estructuras 3D de la proteína o el ligando?
Sí, es preferible proporcionar los IDs de PDB o archivos SDF. Sin embargo, si no los tienes, envíame un mensaje primero para ayudarte con la recuperación o modelado estructural.


