Explorar las categorías
Explora
Fiverr Pro
Español
$
USD
Sobre este servicio
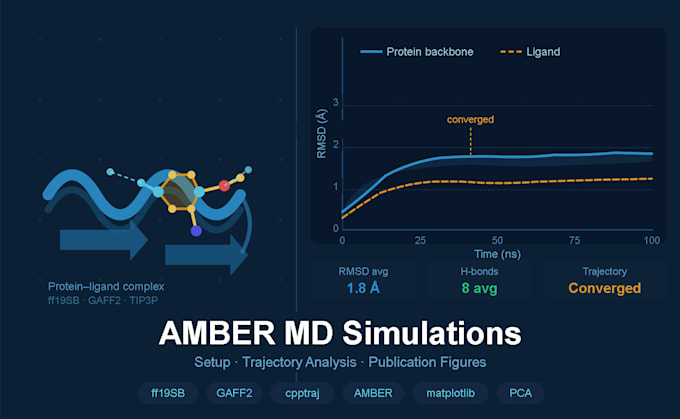
¿Estás trabajando en un sistema proteína-ligando y necesitas simulaciones de dinámica molecular confiables y bien analizadas para tu investigación o publicación? Ofrezco servicios completos de simulación MD con AMBER, desde la preparación del sistema hasta figuras listas para publicación, usando el mismo proceso que en investigaciones revisadas por pares en descubrimiento de fármacos computacional.
Soy investigador en biología computacional con experiencia práctica en AMBER/AmberTools, cpptraj y flujos de trabajo de análisis en Python. Mi trabajo ha apoyado directamente manuscritos enviados a revistas revisadas por pares en farmacología computacional.
Lo que entrego
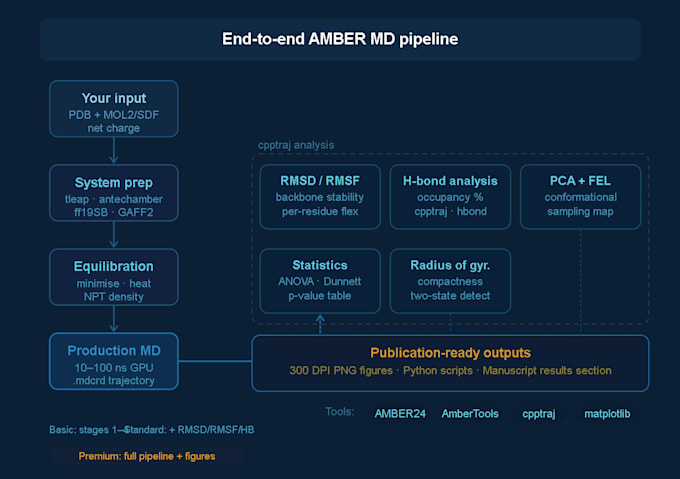
Configuración del sistema
Preparación de la estructura de la proteína y asignación del estado de protonación
Parametrización del ligando usando antechamber
Solvatación (agua TIP3P o OPC), adición de contraiones
Asignación del campo de fuerza: ff19SB (proteína), GAFF2 (molécula pequeña)
Minimización, calentamiento y equilbrio NPT/NVT
Ejecución de MD de producción
Trayectorias de 10, 50 o 100 ns (dependiendo del paquete)
Archivos completos de trayectoria .mdcrd entregados
Análisis de trayectoria (estándar y premium)
Figuras completas listas para publicaciones
¿Por qué trabajar conmigo?
Experiencia real en amber, no una herramienta en línea ni un envoltorio en la nube.